Silica aerogels are interesting materials, composed of silica nanoparticles linked together in a highly porous network. These materials perform exceptionally well as thermal insulators, most notably in spacecraft but also increasingly more in the building insulation market, and as adsorbents for organic pollutants in contaminated water. One major hurdle of producing and handling silica aerogels is their extremely high brittleness.
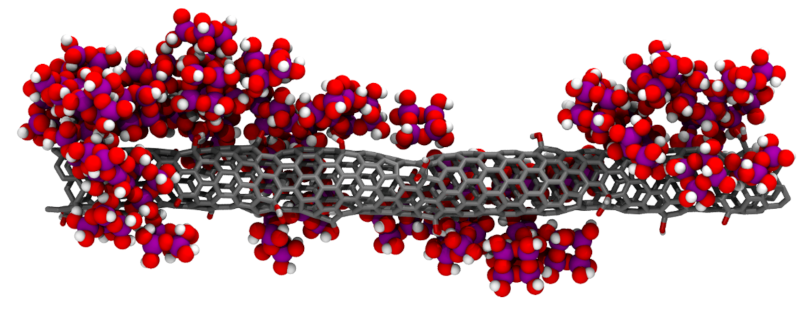
This issue can be alleviated by adding reinforcing materials to the aerogel matrix, such as polymers, fibers, and, more recently, carbon nanotubes (CNT). Particularly for the latter case, the resulting composites not only are much more resistant but can also feature improved thermal insulation and adsorption abilities, among other benefits.
The main challenge in producing silica-CNT composites lies in finding the optimal way to bind the silica aerogel particles to the CNTs, such as to maximize the material’s resistance and potentially enhance other properties. Furthermore, understanding how the silica aerogel is being formed around the CNT, from condensation reactions between smaller precursor molecules, is crucial to design better composite materials. This is a type of problem that can be found in the realm of chemical product engineering that calls for the use of advanced molecular modeling and simulation tools, supported by high performance computing.
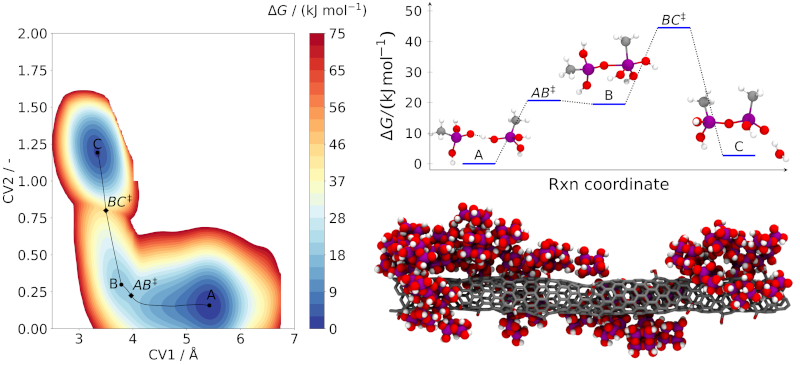
As part of a PhD thesis being conducted at Chemical Process Engineering and Forest Products Research Centre (CIEPQPF) of the Department of Chemical Engineering (DEQ) of the University of Coimbra (UC), simulations using models based on both classical and ab initio molecular dynamics are allowing to obtain a clear and reliable picture of the mechanism of condensation reactions that leads to the formation of silica aerogel particles in different conditions, as well as the energetics of such reactions.
This approach is also being used to explore the chemical and physical interactions between silica particles and CNTs possessing different surface chemical groups. The use of the Navigator Cluster at the Laboratório de Computação Avançada (LCA) of UC is crucial in this type of simulations, which are very demanding in terms of computer resources (both memory and CPU), especially for large systems such as those being studied in this work.