@MoreiraLAB www.moreiralab.com
O nosso projeto, VIRUSHOSTAI – DSAIPA/DS/0118/2020, é um bom exemplo da vantagem de utilizar a Computação de Alto Desempenho, pois envolve simulações de dinâmica molecular de proteínas, bem como os mais recentes algoritmos de aprendizagem profunda. Estamos a seguir uma abordagem baseada em rede para prever novas ligações entre vírus e os seus hospedeiros, essencial para o estabelecimento de uma nova medicina de precisão molecular baseada em informação. Este projeto foi premiado no âmbito do quadro: “AI 4 COVID-19″: Data Science and Artificial Intelligence in the Public Administration to strengthen the fight against COVID-19 and future pandemics – 2020”.
Infelizmente, todos estamos familiarizados com a Síndrome Respiratória Aguda Grave CoronaVirus-2 (SARS-CoV-2), um membro da família Coronaviridae, responsável pela doença de COronaVIrus (COVID-19), uma pandemia que assolou o mundo nos últimos dois anos. Desde o seu primeiro relatório, em dezembro de 2019, em Wuhan, China, foram notificados mais de 355 milhões de casos de infeção e mais de 5,6 milhões de mortes em todo o mundo (dados de 25 de janeiro de 2022).
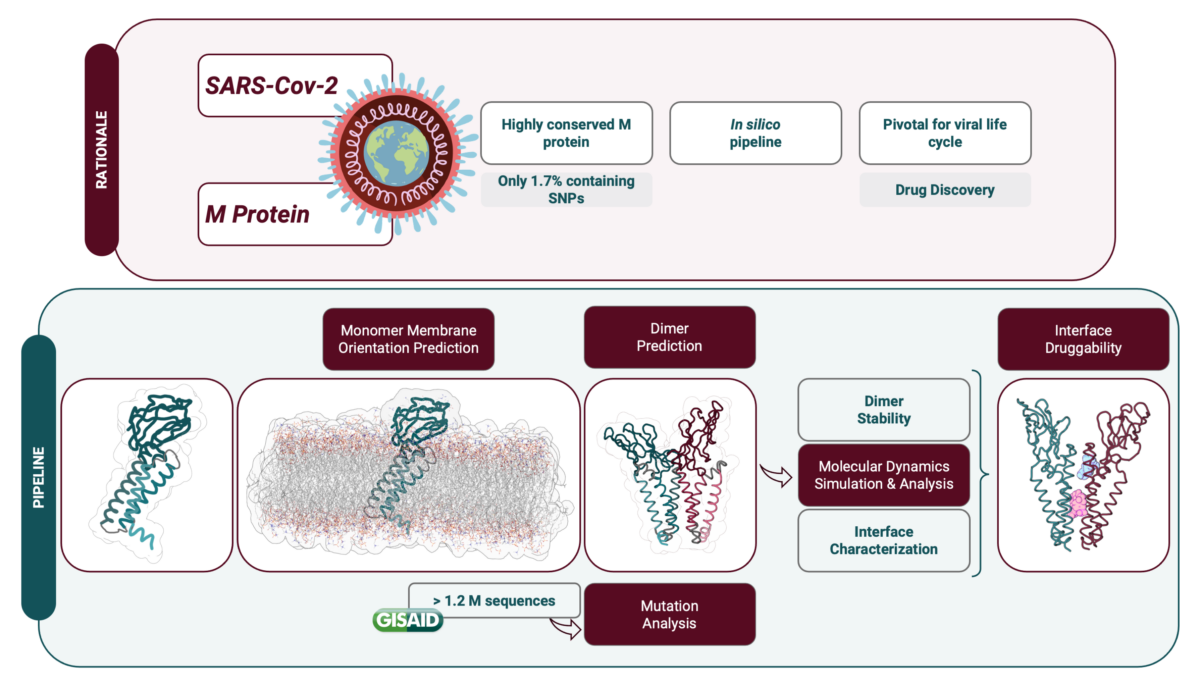
Uma das proteínas estruturais mais conservadas da SARS-CoV-2 é a proteína Membrana (M), uma vez que tem uma taxa de mutação menor e partilha semelhanças estruturais e funcionais com proteínas M de outros coronavírus. Esta proteína tem várias funções e, apesar de algumas ainda não serem completamente compreendidas, desempenha um papel importante na infeção viral. De facto, a proteína M é um ator central na formação do virião através de interações consigo mesma (aquando da formação de um homodímero) e com outras proteínas estruturais e lípidos.
A estrutura da proteína M ainda não foi determinada experimentalmente, mas a empresa DeepMind através do uso do algoritmo AlphaFold publicou, logo no início da pandemia, a previsão da sua estrutura, que serviu de base para o nosso trabalho. Assim, desenvolvemos um protocolo computacional detalhado para prever a estrutura homodimérica da proteína M e a orientação correta da membrana, bem como para analisar os efeitos estruturais e dinâmicos das mutações relatadas em todo o mundo. Além disso, previmos o impacto das mutações em potenciais resíduos de ligação a fármacos, especificamente na área da interface do dímero.
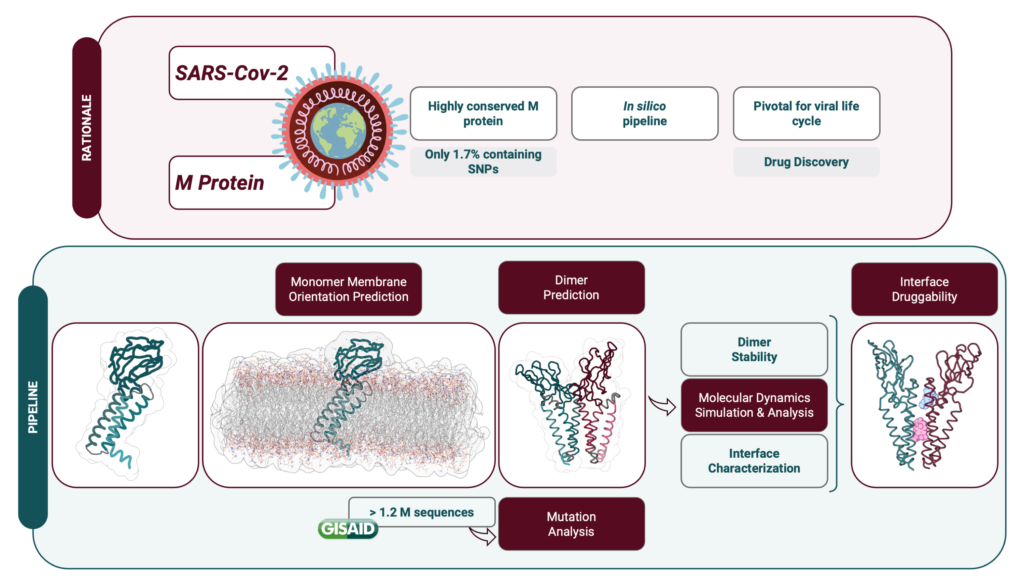
Neste estudo, alargámos claramente o conhecimento sobre a relação estrutura-função da proteína M e previmos novos locais de ligação a fármacos, revelando o potencial terapêutico deste alvo biológico. Estamos agora a utilizar algoritmos computacionais dispendiosos de inteligência artificial para construir uma abordagem que considera as doenças infeciosas no quadro de redes complexas emergentes da integração de informação de vários níveis ómicos para identificar novas possíveis interações antivirais de fármacos-proteínas. Devido à capacidade informática necessária, a utilização do Cluster Navigator – Laboratório de Computação Avançada (LCA) da Universidade de Coimbra, será fundamental para a prossecução dos nossos objetivos.